FDA 510(k) Pathway and Opportunities Introduction
The US is becoming a priority market for medical device development more than ever before. And most of current and new medical projects/innovations prioritize a submission through the American Food and Drug Administration (FDA) as a priority, rather than going straight for the EU market. There are some reasons for this, including the opportunity to address a larger market in the USA compared to EU, thus increasing business potential, and other factors like the reimbursement policies, and the overall regulatory efficiency of FDA.
FDA Regulations for Orthopedic Implants | FDA example
The FDA employs a risk-based approach to regulate the introduction of medical devices, including orthopedic implants, into the US market. This process is designed to ensure both the safety and efficacy of medical devices, while fostering innovation within the industry. The market approval process involves stringent design control protocols, rigorous change control procedures, and adherence to overarching FDA regulations (fda 21 cfr 820). BAAT Medical plays a critical role as a consultant in helping manufacturers navigate these complex processes, starting from the definition of the regulatory pathway, and the identification of which class the medical device falls into.
Class I
Class I devices are “exempt” from the 510(k) process, and only general controls are applicable for their safety and effectiveness evaluation.
Class II
Class II devices are not “exempt” and they must go through the 510(k) process, meaning one should prove their substantial equivalency (SE) to a predicate device (i.e. an existing, US-marketed device with the same intended use).
Class II
Class II devices whose equivalency cannot be demonstrated fall within the De Novo process. In this case, the FDA establishes a new “device type” along with its classification, regulation, necessary controls (general and special controls), and product code.
Class III
Class III devices usually fall within the PMA (Pre-Market Approval) process, and the applicant must provide Clinical Data to substantiate their safety and effectiveness (see Figure 1).
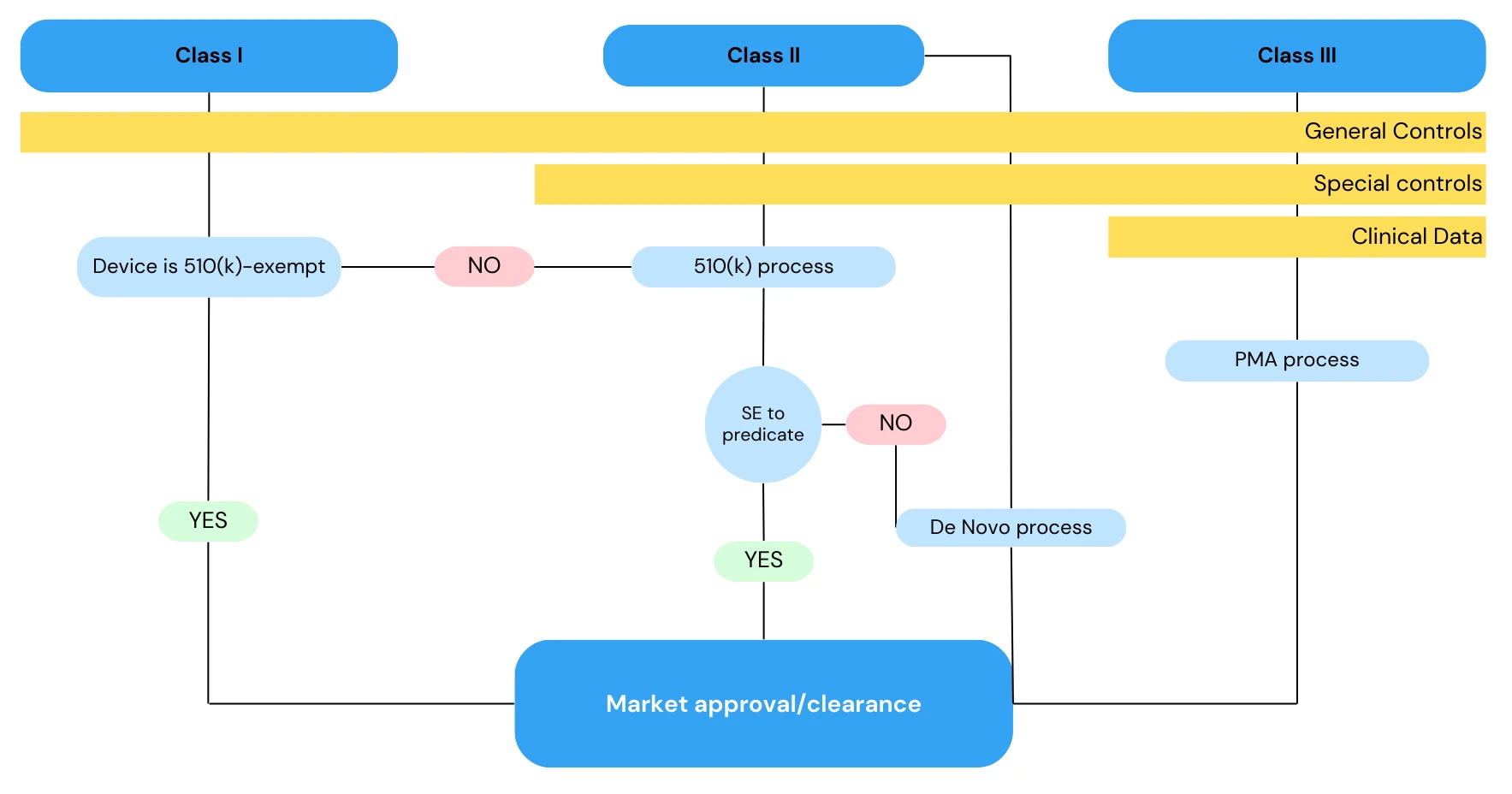
Figure 1. 510k Medical Device Flowchart cfr 820
And how to de-risk your decision?
Here is where the FDA’s regulatory efficiency comes into play, as the agency offers a number of opportunities before (and during) the marketing application for medical devices, including orthopedic implants. For example, the FDA has in place (and recommends) Q-Submission programs that can be initiated to gather feedback on a (future) marketing application. These programs provide interaction with the FDA review team, both in a written and non-written form (e.g., videoconferences). Feedback is provided in a reasonable timeframe: the review time of a Q-Submission is usually 70 calendar days.
Nevertheless, there are ways to speed up the process, provided that certain conditions are met. In fact, the FDA has arranged a number of Special Programs – Breakthrough Device Program, and Safer Technologies Program, to name a few. These programs are aimed at expediting development and acceptance of certain medical devices. They are mainly reserved to products that address debilitating conditions/illnesses, and/or provide a safer or more efficient way of diagnosing medical problems.
Eligible Medical Devices 510k, De Novo, PMA submissions
Medical devices are eligible for such programs only if they meet specific conditions, and if they are subject to premarket approval applications (PMA), premarket notification [510k)], or requests for De Novo classification request.
In these cases, the FDA would prioritize submissions related to medical devices that are granted this kind of “status”. The interaction is quicker, prompt feedback is provided on the development process, all to ensure that a medical device that answers important clinical needs would be available on the market in the least amount of time.
BAAT Medical has built up extensive experience over the years through its consultancy on the dynamic of the FDA processes and programs, with particular focus on orthopedic implants. We strive to de-risk the regulatory pathway for our customers’ medical devices, to make sure that we are always conscious of the effort needed to their safety and performance, and to smoothen the US marketing approval.